Current Research Projects
Structural modeling of molecular assemblies lies at the heart of understanding molecular interactions and biological function. We present a method for docking protein molecules and elucidating native-like structures of protein dimers using geometric hashing to ensure the feasibility of searching the combined conformational space of dimeric structures.
The search space is narrowed by focusing the sought rigid-body transformations around surface areas with evolutionary-conserved amino-acids. Recent analysis of protein complexes reveals that
However, computational docking methods are often not accurate and their results need to be further refined to improve interface packing. We introduce a novel refinement method that incorporates evolutionary information by employing an energy function containing Evolutionary Trace (ET)-based scoring function, which also takes shape complementarity, electrostatic and Van der Waals interactions into account. Our refinement method is able to produce structures with better RMSDs with respect to the known complexes and lower energies than those initial docked structures.
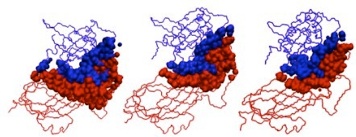
Initial docked solution, the refined version, and the native structure for 1DS6, respectively.
many functional interfaces are significantly conserved throughout evolution. Our results show that focusing the search around evolutionary-conserved interfaces results in lower lRMSDs.
Advisor: Prof. Nurit Haspel
Research Lab: Computational Biology Lab
Collaborators: Prof. Amarda Shehu and Irina Hasmi, GMU
DOCKING AND REFINING PROTEIN COMPLEXES USING EVOLUTIONARY INFORMATION
Some regions in proteins play a critical role in determining their structure and function. Examples include flexible regions such as hinges which allow domain motions, and highly conserved functional interfaces which allow interactions with other proteins. Detecting these regions facilitates the analysis and simulation of protein rigidity and conformational changes and aids in characterizing protein-protein binding.
DETECTING CRITICAL RESIDUES IN PROTEINS
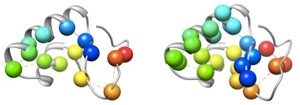
Known critical residues and detected critical residues of Crambin, respectively.
performs rigidity analysis to find rigid clusters of amino acids in a protein and the other method uses evolutionary conservation to find functional interfaces in proteins. We combined the results of both methods as features of a Support Vector Machine, which is trained with a dataset of proteins with experimentally known critical residues. The results show that the combination of the two methods can detect the vast majority of critical residues in tested proteins.
We conduct an analysis of critical residues in proteins using a combination of two complementary methods. One method
Advisor: Prof. Nurit Haspel
Research Lab: Computational Biology Lab
Collaborators: Prof. Filip Jagodzinski, CWU
A major challenge for protein-protein docking problem is to accurately discriminate native-like structures among a large set of solution candidates. Scoring functions are used to achieve this goal. The protein docking community agrees on the existence of a relationship between various favorable intermolecular interactions (e.g. Van der Waals, electrostatic, desolvation forces, etc.) and the similarity of a conformation to its native structure. Therefore, different docking algorithms often formulate their scoring functions as a weighted sum of selected terms and calibrate their weights against a specific training data. However, the exact form of the relationship between these features and a structure’s similarity to its native form is unknown. This significantly reduces the accuracy of the conventional scoring functions.
In this research, we approximate this complex relationship using a novel machine learning approach. We propose a
PREDICTING STRUCTURE SIMILARITY OF DOCKED PROTEIN COMPLEXES USING A MACHINE LEARNING APPROACH
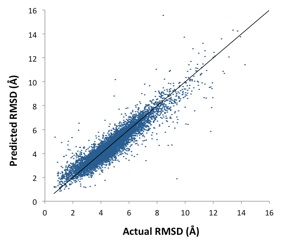
backpropagation neural network to predict the RMSD value of a given refinement candidate with respect to its native conformation. Then, using the predicted RMSD values, we rank the candidate structures.
Advisor: Prof. Nurit Haspel
Research Lab: Computational Biology Lab
Collaborators: Prof. Marc Pomplun, UMB