Research Topics (constantly under construction)
Conformational search of medium resolution proteins
Many large protein complexes undergo extensive conformational
changes as part of their functionality.
Tracing these changes is important for understanding the way these proteins function. It is not always possible to obtain a high resolution
structure for very large complexes. Cryo-Electron microscopy (Cryo-EM) is a powerful tool that enables the representation of large macromolecular
structures at a medium resolution level (6--9\AA).
While many conformational search methods explore the motions of atomic resolution protein structures, little has been done to handle the abundance of medium
resolution data available. Traditional conformational search methods are
impractical
for very large complexes due to the amount of computational time involved.
Moreover, they cannot be applied to medium resolution data
where structural information may be partial or missing.
To address this problem, we propose a novel computational methodology to efficiently trace
the conformational changes in biological macromolecules represented
as medium resolution structures. We develop and apply a search method from robotics to
structural information obtained from Cryo-EM.
Our method is unique in its ability to conduct a computationally tractable search,
using approximate data to obtain approximate but reliable
results. The pathways obtained by this method can be useful in understanding
protein motion and functionality. To provide a baseline test for our method, we
tested it on Adenylate Kinase and the GroEL monomer. We show that we can
produce low energy conformational pathways with accuracy well below
the structure's resolution level. This method is a promising first
step towards exploring the conformational motion of even larger
complexes.
This work is done in collaboration with Dr. Wah Chiu from Baylor College of Medicine.
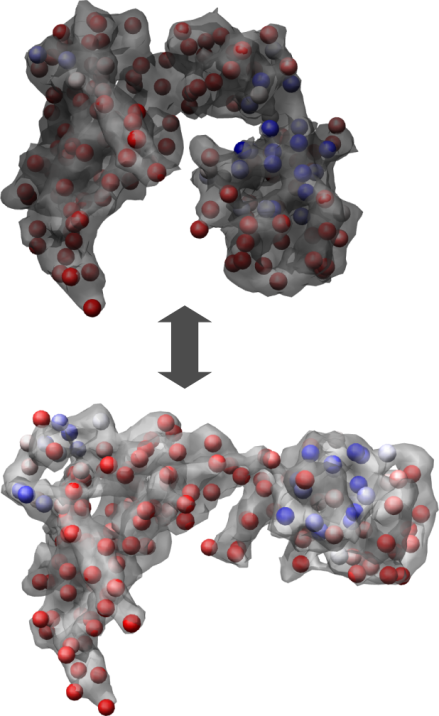
A cryo-EM model of the GroEL monomer (top) and the GroEL-GroES-ADP7 monomer (bottom).
Characterizing the binding properties of the C3d/Efb-C complex
The C3-inhibitory domain of Staphylococcus aureus extracellular fibrinogen-binding protein (Efb-C) defines a novel three-helix bundle motif that regulates complement activation. Previous crystallographic studies of Efb-C bound to its cognate sub-domain of human C3 (C3d) identified Arg-131 and Asn-138 of Efb-C as key residues for its activity. In order to characterize more completely the physical and chemical driving forces behind this important interaction, we employed in this study a combination of structural, biophysical, and computational methods to analyze the interaction of C3d with Efb-C and the single point mutants R131A and N138A. Our results show that while these mutations do not drastically affect the structure of the Efb-C/C3d recognition complex, they have significant adverse effects on both the thermodynamic kinetic profiles of the resulting complexes. We also characterized other key interactions along the Efb-C/C3d binding interface and found an intricate network of salt bridges and hydrogen bonds that anchor Efb-C to C3d, resulting in its potent complement inhibitory properties.
We are currently developing a multi-scale search technique to explore the local conformational dynamics of proteins using a combination of a fast robotics based structural modeling algorithm, MD simulations and dimensionality reduction methods. The method showed promising results in exploring the conformational space of the C3d/Efb-C complex and mutants. The method showed an ability to capture a larger portion of the conformational landscape of the complex than the MD study. The results are currently being tested against experimental data. Part of the computational work was carried out using the TeraGrid high-performance computing resources.
This work is done in collaboration with Dr. John Lambris, UPenn and Dr. Brian Geisbrecht, UMKC.
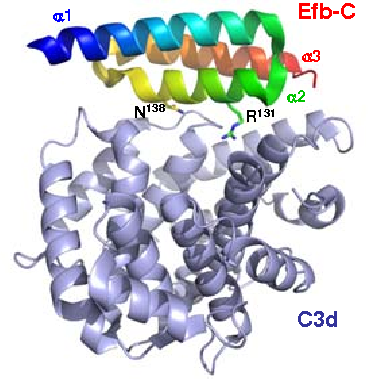
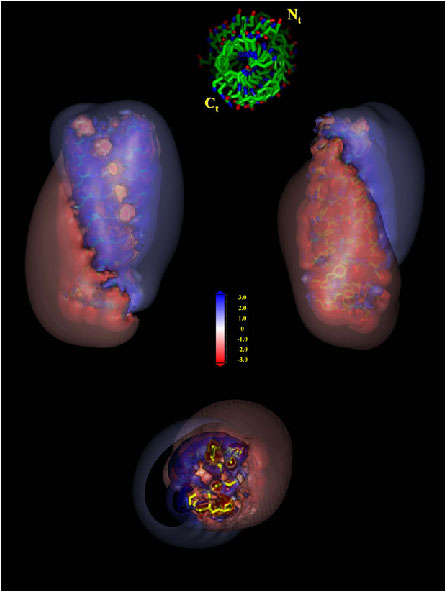
The C3d/Efb-C complex. Residues R131 and N138 on Efb-C are indicated.
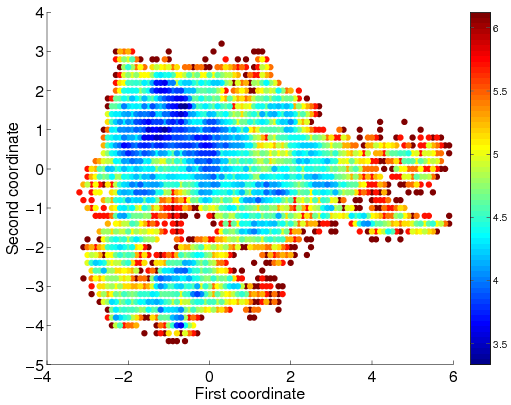
The C3d/Efb-C low energy surface as obtained by multi scale sampling. The surface is colored according to free energy where shades of red indicate high energy regions and shades of blue indicate low energy regions.
Nano-design of novel structures based on protein motifs
In this work we devise an approch for designing self-assembled nanostructures from naturally occurring building block segments obtained from native protein structures. We focus on structural motifs from left-handed beta-helical proteins. We selected 17 motifs. Copies of each of the motifs are stacked one atop the other. The obtained structures were simulated for long periods by using Molecular Dynamics to test their ability to retain their organization over time. We observed that a structural model based on the self-assembly of a motif from E. coli galactoside acetyltransferase (PDB 1krr) produced a very stable tube. We studied the interactions that help maintain the conformational stability of the systems, focusing on the role of specific amino acids at specific positions. Analysis of these systems and a mutational study of selected candidates revealed that the presence of proline and glycine residues in the loops of beta-helical structures greatly enhances the structural stability of the systems. Later, we modified this model by changing the charge distribution in the inner core of the system and testing the effect of this change on the structural arrangement of the construct. Our results demonstrate that it is possible to generate the proper conditions for charge transfer inside nanotubes based on assemblies of krr1 segment. The electronic transfer would be achieved by introducing different histidine ionization states in selected positions of the internal core of the construct, in addition to specific mutations with charged amino acids that altogether will allow the formation of coherent networks of aromatic ring stacking, salt-bridges, and hydrogen bonds.
This work was done in collaboration with Dr. David Zanuy, National Cancer Institute.
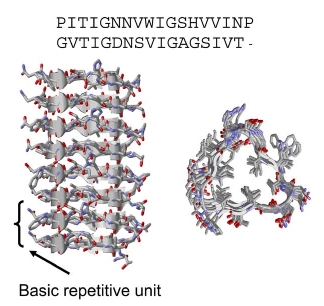
Residues 131–165 of Galactoside Acetyltransferase. Top: The sequence alignment of the rungs of the repeat.
Computational modeling of short amyloid structures
Experimentally, the human calcitonin hormone (hCT) can form highly stable amyloid protofibrils. Further, a peptide consisting of hCT residues 15-19, DFNKF, was shown to create highly ordered fibrils, similar to those formed by the entire hormone sequence. We have modeled the DFNKF protofibril, using molecular dynamics simulations. We tested the stabilities of single sheet and of various multi sheet models. Remarkably, our most ordered and stable model consists of a parallel-stranded, single beta-sheet with a relatively insignificant hydrophobic core. We investigate the chemical and physical interactions responsible for the high structural organization of this single beta-sheet amyloid fibril. We observe that the most important chemical interactions contributing to the stability of the DFNKF organization are electrostatic, specifically between the Lys and the C terminus, between the Asp and N terminus, and a hydrogen bond network between the Asn side-chains of adjacent strands. Additionally, we observe hydrophobic and aromatic pi stacking interactions. We further simulated truncated filaments, FNKF and DFNK. Our tetra-peptide mutant simulations assume models similar to the penta-peptide. Experimentally, the FNKF does not create fibrils while DFNK does, albeit short and less ordered than DFNKF. In the simulations, the FNKF system was less stable than the DFNK and DFNKF. DFNK also lost many of its original interactions becoming less organized, however, many contacts were maintained. Thus, our results emphasize the role played by specific amino acid interactions. To further study specific interactions, we have mutated the penta-peptide, simulating DANKF, DFNKA and EFNKF. Here we describe the model, its relationship to experiment and its implications to amyloid organization.
This work was done in collaboration with Dr. David Zanuy, National Cancer Institute.
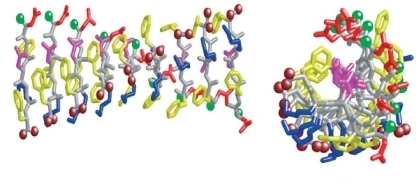
The suggested model for the DFNKF proto-fibril
Charge distribution for the DFNKF proto-fibril
A computational study of protein structures based on the building block folding model
This study is based on the previously presented building block folding model. The model postulates that protein folding is a hierarchical top-down process. The basic unit from which a fold is constructed, referred to as a hydrophobic folding unit, is the outcome of combinatorial assembly of a set of "building blocks." Results obtained by the computational cutting procedure yield fragments that are in agreement with those obtained experimentally by limited proteolysis. Here we show that as expected, proteins from the same family give very similar building blocks. However, different proteins can also give building blocks that are similar in structure. In such cases the building blocks differ in sequence, stability, contacts with other building blocks, and in their 3D locations in the protein structure. This result, which we have repeatedly observed in many cases, leads us to conclude that while a building block is influenced by its environment, nevertheless, it can be viewed as a stand-alone unit. For small-sized building blocks existing in multiple conformations, interactions with sister building blocks in the protein will increase the population time of the native conformer. With this conclusion in hand, it is possible to develop an algorithm that predicts the building block assignment of a protein sequence whose structure is unknown. Toward this goal, we have created sequentially nonredundant databases of building block sequences. A protein sequence can be aligned against these, in order to be matched to a set of potential building blocks. In a later study we used the building block library to develop an algorithm that selects optimal combinations to "cover" the protein sequence. The input is a target protein sequence and a library of non-redundant building block fragments generated by cutting all protein structures. The output is a set of cutout fragments.
Related publications